
|
CCP4i: Graphical User Interface |
| MR Tutorial Bath - Pepsin case study |
 |
BACK TO INDEX |  |
Convert the reflection data file into the internal format used by the AMORE program.
# amore HKLIN hexpep HKLPCK0 hexpep.hkl <<EOD TITLE ** packing h k l F for crystal ** SORT LABIN FP=FOHEXPEP SIGFP=SIGHEXPEP EOD
Compute a molecular Fourier transform for use in structure factor calculation by interpolation.
# amore XYZIN1 penpep XYZOUT1 penpep_model table1 penpep.tab <<EOD TITLE Produce table for penicillopepsin model. TABFUN NOROTA ! No rotate for comparison. CRYST CELL 67.4 67.4 290.1 90 90 120 MODEL 1 BREP 10 SAMPLE 1 RESO 2.8 ! Beyond native resolution for interpolation. EOD
Note that the steps in the procedure need not be run as separate jobs as illustrated in the two previous examples; this has only been done in order to simplify explanation of the individual steps. The next example shows several steps being run sequentially in a single job.
Compute structure factor amplitudes for the search model in space group P1 in a large rectangular cell.
Compute spherical harmonic coefficients for the target and model Pattersons.
Compute the cross-RF.
#
amore HKLPCK0 hexpep.hkl table1 penpep.tab HKLPCK1 penpep.hkl \
CLMN0 hexpep.clmn CLMN1 penpep.clmn MAPOUT hexpep_rotfun <<EOD
ROTFUN
TITLE Generate HKLPCK1 from penicillopepsin model.
GENER 1 RESO 20 3 CELL 83 82 92
CLMN CRYST RESO 20 3 SHARP -10 SPHERE 30
CLMN MODEL 1 RESO 20 3 SHARP -10 SPHERE 30
ROTATE CROSS MODEL 1 BMAX 90 PKLIM .25 NPIC 20 ! Beta = 0-90
EOD
\rm hexpep_rotfun.map
Radius = 25Å. B = -10Ų.
| a | b | g | S | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SOLUTIONRC | 1 | 33.08 | 78.55 | 36.38 | 0.00000 | 0.00000 | 0.00000 | 7.1 | 0.0 |
| SOLUTIONRC | 1 | 35.02 | 87.24 | 129.83 | 0.00000 | 0.00000 | 0.00000 | 6.7 | 0.0 |
| SOLUTIONRC | 1 | 24.00 | 69.00 | 263.50 | 0.00000 | 0.00000 | 0.00000 | 6.7 | 0.0 |
| SOLUTIONRC | 1 | 28.36 | 78.25 | 315.14 | 0.00000 | 0.00000 | 0.00000 | 6.5 | 0.0 |
| SOLUTIONRC | 1 | 41.19 | 29.92 | 23.72 | 0.00000 | 0.00000 | 0.00000 | 6.4 | 0.0 |
| SOLUTIONRC | 1 | 31.65 | 39.80 | 256.04 | 0.00000 | 0.00000 | 0.00000 | 6.4 | 0.0 |
| SOLUTIONRC | 1 | 18.50 | 23.34 | 271.50 | 0.00000 | 0.00000 | 0.00000 | 6.4 | 0.0 |
Radius = 30Å. B = 0.
| SOLUTIONRC | 1 | 30.14 | 37.23 | 256.28 | 0.00000 | 0.00000 | 0.00000 | 5.6 | 0.0 |
| SOLUTIONRC | 1 | 35.25 | 30.37 | 244.50 | 0.00000 | 0.00000 | 0.00000 | 5.5 | 0.0 |
| SOLUTIONRC | 1 | 41.00 | 28.32 | 19.00 | 0.00000 | 0.00000 | 0.00000 | 5.4 | 0.0 |
|---|---|---|---|---|---|---|---|---|---|
| SOLUTIONRC | 1 | 32.75 | 85.70 | 129.88 | 0.00000 | 0.00000 | 0.00000 | 5.0 | 0.0 |
| SOLUTIONRC | 1 | 32.99 | 78.33 | 35.83 | 0.00000 | 0.00000 | 0.00000 | 4.9 | 0.0 |
Radius = 30Å. B = -10Ų.
| SOLUTIONRC | 1 | 44.00 | 28.35 | 16.00 | 0.00000 | 0.00000 | 0.00000 | 5.1 | 0.0 |
|---|---|---|---|---|---|---|---|---|---|
| SOLUTIONRC | 1 | 33.94 | 86.23 | 129.99 | 0.00000 | 0.00000 | 0.00000 | 5.0 | 0.0 |
| SOLUTIONRC | 1 | 34.82 | 30.43 | 244.68 | 0.00000 | 0.00000 | 0.00000 | 4.9 | 0.0 |
The table below shows values of the signal/noise ratio DS/s for the correct peak relative to the highest background peak. A "-" indicates that the correct peak was not present in the top 20 peak list.
| B | |||
|---|---|---|---|
| Radius/Š| 0 | -10Ų | -20Ų |
| 15 | - | - | - |
| 20 | - | - | - |
| 25 | -0.44 | -0.36 | -0.33 |
| 30 | -0.13 | 0.07 | -0.07 |
| 35 | - | -1.05 | -0.83 |
| 40 | - | - | -0.55 |
Note: Other combinations of resolution limits (8-3Å, 20-3.5Å, 8-3.5Å), radius or B parameter did not give correct peak in list.
Modify the PDB header in the coordinate file.
# pdbset XYZIN penpep XYZOUT penpep_rfcell <<EOD SPACEG P1 CELL 83 82 92 EOD
Compute structure factor amplitudes in space group P1 for the search model in a large rectangular cell.
# # Structure factors for penicillopepsin model in RF cell. # sfall XYZIN penpep_rfcell HKLOUT penpep_rfcell <<EOD MODE SFCALC XYZIN SFSG 1 SYMM 1 RESO 20 3 BADD 10 LABOUT PHIC=PC EOD
Normalise the observed and calculated amplitudes.
# ecalc HKLIN hexpep HKLOUT hexpep_ecalc <<EOD TITLE ** Ecalc for target crystal** LABIN FP=FOHEXPEP SIGFP=SIGHEXPEP EOD ecalc HKLIN penpep_rfcell HKLOUT penpep_ecalc <<EOD TITLE ** Ecalc for model** LABIN FP=FC EOD
Convert the normalised observed and calculated reflection data files into the internal format used by the AMORE program.
# amore HKLIN hexpep_ecalc HKLPCK0 hexpep_ecalc.hkl <<EOD TITLE ** packing h k l E for target crystal ** SORT LABIN FP=E SIGFP=E EOD amore HKLIN penpep_ecalc HKLPCK0 penpep_ecalc.hkl <<EOD TITLE ** packing h k l E for model ** SORT LABIN FP=E SIGFP=E EOD
Compute spherical harmonic coefficients for the target and model Pattersons.
Compute the cross-RF.
#
amore HKLPCK0 hexpep_ecalc.hkl HKLPCK1 penpep_ecalc.hkl \
CLMN0 hexpep_ecalc.clmn CLMN1 penpep_ecalc.clmn \
MAPOUT hexpep_ecalc_rotfun <<EOD
ROTFUN
TITLE Rotation function with E's..
CLMN CRYST RESO 20 3 SPHERE 25
CLMN MODEL 1 RESO 20 3 SPHERE 25
ROTATE CROSS MODEL 1 BMAX 90 PKLIM .25 NPIC 20
EOD
\rm hexpep_ecalc_rotfun.map
| a | b | g | S | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SOLUTIONRC | 1 | 43.50 | 21.00 | 14.00 | 0.00000 | 0.00000 | 0.00000 | 7.3 | 0.0 |
| SOLUTIONRC | 1 | 42.17 | 29.66 | 26.30 | 0.00000 | 0.00000 | 0.00000 | 6.5 | 0.0 |
| SOLUTIONRC | 1 | 52.38 | 54.25 | 128.35 | 0.00000 | 0.00000 | 0.00000 | 6.1 | 0.0 |
| SOLUTIONRC | 1 | 32.72 | 58.97 | 66.17 | 0.00000 | 0.00000 | 0.00000 | 6.1 | 0.0 |
Note that 2 peaks separated by a small rotation angle (about 14o) are observed in the Rotation function based on E's, whereas only a single peak was observed when F's were used; thus normalisation produces a Rotation function map with a much higher effective resolution. The splitting is due to hinge-bending giving differing orientations of the N- and C-terminal domains of the aspartic proteinase. In hindsight it might have been better to separate the model into its constituent domains, and treat it as a 2-molecule problem.
The table below shows values of DS/s.
| Resolution limits | ||
|---|---|---|
| Radius/Å | 20-3Å | 20-3.5Å |
| 15 | 0.09 | -0.73 |
| 20 | 0.31 | -0.45 |
| 25 | 0.73 | - |
| 30 | 0.32 | -1.03 |
| 35 | 0.61 | -0.26 |
| 40 | 0.62 | -0.10 |
The table below shows the effect on the signal/noise ratio of the RF of omitting the largest intensities (resolution limits: 20-2Å).
| Coeff. | B/Ų | # omitted (%) | ||
|---|---|---|---|---|
| none | 66 (1.1) | 171 (2.9) | ||
| F | 0 | 4.2 | -0.6 | - |
| F | -20 | 6.9 | 3.9 | 1.6 |
| E | 0 | 6.6 | 4.9 | 2.8 |
Note: Negative signal/noise means that the correct peak is lost in the noise!
For the best RF solutions, compute the TF's.
# amore HKLPCK0 hexpep.hkl table1 penpep.tab MAPOUT hexpep_trafun.map <<EOD TRAFUN CB RESO 20 3.5 TITLE Translation function SYMM P6522 SOLUTIONRC 1 30.14 37.23 256.28 0.00000 0.00000 0.00000 5.6 0.0 SOLUTIONRC 1 35.25 30.37 244.50 0.00000 0.00000 0.00000 5.5 0.0 SOLUTIONRC 1 41.00 28.32 19.00 0.00000 0.00000 0.00000 5.4 0.0 SOLUTIONRC 1 32.75 85.70 129.88 0.00000 0.00000 0.00000 5.0 0.0 SOLUTIONRC 1 32.99 78.33 35.83 0.00000 0.00000 0.00000 4.9 0.0 EOD
The table below is for the correct RF solution.
| Resolution limits |
Correlation coefficient | ||
|---|---|---|---|
| DS/s | Correct | Background | |
| 20-4Å | 1.69 | 0.212 | 0.153 |
| 20-3.5Å | 2.09 | 0.210 | 0.169 |
| 8-3.5Å | 0.81 | 0.172 | 0.153 |
| 20-3Å | 1.62 | 0.264 | 0.226 |
No NCS, so this step not applicable.
For the best TF solutions, do rigid-body refinements and choose the solution with the highest correlation coefficient.
# amore HKLPCK0 hexpep.hkl table1 penpep.tab <<EOD FITFUN RESO 20 3.5 TITLE Rigid body refinement SYMM P6522 REFSOL AL BE GA X Y Z BF SOLUT_1 1 43.50 21.00 14.00 0.0772 0.6903 0.2988 19.5 53.0 1 SOLUT_1 1 43.50 21.00 14.00 0.5903 0.1203 0.3246 13.3 54.4 2 SOLUT_1 1 43.50 21.00 14.00 0.8148 0.4523 0.3953 12.7 54.9 9 SOLUT_2 1 42.17 29.66 26.30 0.0719 0.2201 0.3324 13.4 54.7 4 SOLUT_2 1 42.17 29.66 26.30 0.0789 0.2299 0.4226 13.1 54.9 2 SOLUT_3 1 52.38 54.25 128.35 0.6740 0.9740 0.4168 13.9 54.9 9 SOLUT_3 1 52.38 54.25 128.35 0.4894 0.9219 0.2713 12.9 55.4 2 SOLUT_4 1 32.72 58.97 66.17 0.0550 0.0375 0.0206 12.9 55.1 1 SOLUT_5 1 38.50 59.00 65.69 0.9230 0.1232 0.2007 13.5 53.8 1 SOLUT_5 1 38.50 59.00 65.69 0.9197 0.1238 0.0890 12.5 54.8 2 EOD
INITIAL POSITIONS
1 43.500 21.000 14.000 0.077200 0.690300 0.298800 19.500 53.000
FINAL POSITIONS
SOLUTIONF 1 43.58 22.67 15.07 0.07790 0.69162 0.29897 26.6 51.3
INITIAL POSITIONS
1 43.500 21.000 14.000 0.590300 0.120300 0.324600 13.900 54.900
FINAL POSITIONS
SOLUTIONF 1 45.54 21.02 12.42 0.59161 0.12234 0.32435 18.3 54.4
INITIAL POSITIONS
1 43.500 21.000 14.000 0.814800 0.452300 0.395300 13.500 53.800
FINAL POSITIONS
SOLUTIONF 1 42.93 21.40 14.83 0.81292 0.44990 0.39566 18.3 53.3
| Correlation coefficient | R - factor | |||
|---|---|---|---|---|
| Solution | Before | After | Before | After |
| 1 | 0.195 | 0.266 | 0.530 | 0.513 |
| 2 | 0.139 | 0.183 | 0.549 | 0.544 |
| 3 | 0.135 | 0.183 | 0.538 | 0.533 |
| 4 | 0.134 | 0.183 | 0.547 | 0.543 |
| 5 | 0.133 | 0.181 | 0.544 | 0.541 |
Apply the optimised rotation matrix and translation vector to the model coordinates output by AMORE / TABFUN.
# pdbset XYZIN penpep_model XYZOUT hexpep <<EOD SPACEG P6522 CELL 67.4 67.4 290.1 90 90 120 ROTATE EULER 42.68 26.21 19.27 SHIFT FRACT 0.07790 0.69162 0.29897 EOD
Apply the appropriate rotation matrix, calculated from the Eulerian angles of the peak(s) in the RF, to the model coordinates. Also modify the PDB header in the coordinate file.
# pdbset XYZIN penpep XYZOUT penpep_tfcell <<EOD SPACEG P1 CELL 67.4 67.4 290.1 90 90 120 ROTATE EULER 42.68 26.21 19.27 EOD
Calculate structure factor amplitudes and phases.
# # Structure factors for penicillopepsin model in TF cell. sfall XYZIN penpep_tfcell HKLOUT penpep_tfcell <<EOD MODE SFCALC XYZIN SFSG 1 SYMM 1 RESO 20 3 BADD 10 LABOUT PHIC=PC EOD
Combine the columns for the observed reflection data with those for the calculated molecule into a single file.
# cad HKLIN1 hexpep HKLIN2 penpep_tfcell HKLOUT hexpep_cad <<EOD RESO OVER 20 3 TITLE COMBINING hexagonal pepsin Fobs & penicillopepsin Fcalc. LABIN FILE 1 E1=FOHEXPEP E2=SIGHEXPEP CTYPE FILE 1 E1=F E2=Q LABIN FILE 2 E1=FC E2=PC CTYPE FILE 2 E1=U E2=V LABOUT FILE 2 E1=FC E2=PC END EOD
Compute the Fourier coefficients for the Translation function.
# tffc HKLIN hexpep_cad HKLOUT hexpep_tffc <<EOD TITLE tffc on hexagonal pepsin SPACEG P6522 RESO 20 3.5 LABIN FP=FOHEXPEP SIGFP=SIGHEXPEP EOD
Compute the Fourier transform to get the Translation function map.
# fft HKLIN hexpep_tffc MAPOUT hexpep_tffc <<EOD TITLE FFT of tffc map XYZLIM 0 1 0 1 0 .5 ! Limits for point group 622 GRID 128 128 512 LABIN A=A B=B EOD
Search the Translation function map for peaks; one peak should stand out from the rest with relative signal/noise > 1.
# mapsig MAPIN hexpep_tffc PEAK_LIST hexpep_tffc.pkl <<EOD EOD \rm hexpep_tffc.map
| tx | ty | tz | S/s | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 60 | 24 | 124.0 | 34.2 | 135.3 | 0.560 | 0.968 | 0.268 | 0.264 | 9.22 |
| 2 | 15 | 15 | 81.0 | 77.0 | 136.0 | 0.389 | 0.633 | 0.602 | 0.266 | 6.42 |
| 3 | 29 | 14 | 124.0 | 34.0 | 212.0 | 0.375 | 0.969 | 0.266 | 0.414 | 6.17 |
| 4 | 37 | 17 | 124.0 | 34.0 | 68.0 | 0.372 | 0.969 | 0.266 | 0.133 | 6.12 |
It may be necessary to try an alternative space group (in this case P6122):
# tffc HKLIN hexpep_cad HKLOUT hexpep_tffc <<EOD TITLE tffc on hexagonal pepsin in alternative space group. SPACEG P6122 RESO 20 3.5 LABIN FP=FOHEXPEP SIGFP=SIGHEXPEP EOD if ($status) exit fft HKLIN hexpep_tffc MAPOUT hexpep_tffc <<EOD TITLE FFT of tffc map XYZLIM 0 1 0 1 0 .5 GRID 128 128 512 LABIN A=A B=B EOD if ($status) exit mapsig MAPIN hexpep_tffc PEAK_LIST hexpep_tffc.pkl <<EOD EOD \rm hexpep_tffc.map
| tx | ty | tz | S/s | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 | 8 | 121.0 | 88.4 | 212.6 | 0.340 | 0.945 | 0.691 | 0.415 | 5.76 |
| 2 | 11 | 11 | 28.1 | 24.6 | 221.9 | 0.310 | 0.219 | 0.192 | 0.433 | 5.25 |
| 3 | 12 | 12 | 49.4 | 71.2 | 81.3 | 0.308 | 0.386 | 0.556 | 0.159 | 5.21 |
| 4 | 20 | 10 | 53.9 | 74.2 | 13.5 | 0.305 | 0.421 | 0.579 | 0.026 | 5.15 |
| Resolution | DS/s |
|---|---|
| 20-4Å | 2.61 |
| 20-3.5Å | 2.80 |
| 8-3.5Å | 1.70 |
| 20-3Å | 2.15 |
| ª 20-3.5Å | 0.51 |
ª Alternative space group P6122.
No NCS, so this step not applicable.
Apply the translation vector to the rotated model coordinates.
# pdbset XYZIN penpep_tfcell XYZOUT hexpep <<EOD SPACEG P6522 SHIFT FRACT .968 .268 .264 EOD
The X-PLOR program can also be used to compute the cross-RF and do rigid or segmented body refinement in Patterson space (Patterson Correlation or PC refinement). In theory this should be particularly useful in cases where there is limited flexibility such as hinge-bending. This is common in multi-domain proteins, including the aspartic proteinases.
# mtz2various HKLIN hexpep HKLOUT hexpep.xpl <<EOD OUTPUT XPLOR LABIN FP=FOHEXPEP SIGFP=SIGHEXPEP EOD
#
xplor << \EOD
remarks file generate/generate.inp
remarks Generate structure file and hydrogens for a protein
topology
@topPAR:tophcsdx.pro { Read topology file }
end
parameter
@topPAR:parhcsdx.pro { Read empirical potential }
{ parameter file CHARMM19 }
{ with modifications }
nbonds { This statement specifies the}
atom cdie shift eps=1.0 e14fac=0.4 { nonbonded interaction energy}
cutnb=7.5 ctonnb=6.0 ctofnb=6.5 { options. Note the reduced }
nbxmod="5" vswitch { nonbonding cutoff to save }
end { some CPU time. This statement}
{ overwrites the defaults in the}
{ parameter file. }
end
segment { Generate protein }
name=" " { This name has to match the }
{ four characters in columns 73}
{ through 76 in the coordinate}
{ file, in XPLOR this name is }
{ name is referred to as SEGId.}
chain
@topPAR:toph19.pep { Read peptide bond file }
coordinates @penpep.brk end { interpret coordinate file to}
end { obtain the sequence }
end
{ Sometimes different atom }
vector do (name="O") ( name OT1 ) { names are used... }
vector do (name="OT") ( name OT2 )
vector do (name="CD1") ( name CD and resname ile )
coordinates @penpep.brk end { Here we actually read the }
{ coordinates }
vector do ( B=10 ) ( all ) { B's are zero: reset all to 10}
flags exclude vdw elec end { Do QUICK hydrogen building w/o}
{ vdw and elec terms }
hbuild { This statement builds }
selection=( hydrogen ) { missing hydrogens which are }
phistep="4" { needed for the force field }
end
write coordinates output=penpep_xplor.brk end { Write out coordinates}
write structure output=penpep.psf end { Write out structure file }
stop
\EOD
#
xplor << \EOD
remarks file xtalmr/rotation.inp -- cross rotation function
remarks (model P1 vs crystal)
{===>} structure @penpep.psf end { read structure file}
{===>} coor @penpep_xplor.brk { read coordinates }
{===>} { specify location of Patterson map files}
evaluate ( $p1_map="p1.map" )
evaluate ( $p2_map="p2.map" )
{===>}
evaluate ( $max_vector=30 ) { maximum Patterson vector to be searched}
evaluate ( $m_max_vector=-$max_vector )
xrefin { make Patterson P1 map of model in P1 box}
{===>} { the P1 box has to be larger than twice the}
{ the extend of the model in each direction}
a=100.0 b=100.0 c=120.0 alpha=90.0 beta=90.0 gamma=90.0
symmetry=(x,y,z)
SCATter ( chemical C* )
2.31000 20.8439 1.02000 10.2075 1.58860 .568700 .865000 51.6512 .215600
SCATter ( chemical N* )
12.2126 .005700 3.13220 9.89330 2.01250 28.9975 1.16630 .582600 -11.529
SCATter ( chemical O* )
3.04850 13.2771 2.28680 5.70110 1.54630 .323900 .867000 32.9089 .250800
SCATter ( chemical S* )
6.90530 1.46790 5.20340 22.2151 1.43790 .253600 1.58630 56.1720 .866900
SCATter ( chemical P* )
6.43450 1.90670 4.17910 27.1570 1.78000 0.52600 1.49080 68.1645 1.11490
SCATter ( chemical FE* )
11.1764 4.61470 7.38630 0.30050 3.39480 11.6729 0.07240 38.5566 0.97070
{===>} { allocate sufficient space for the reflections of the P1 box}
nreflections=200000
{===>}
resolution 20 3 { resolution range for P1 box }
generate { generate reflections for P1 box}
method=fft
fft
grid=0.25 { sampling grid for FFT and Patterson map (1/4 high resol.)}
end
update { compute Fcalcs for model in P1 box}
do amplitude (fcalc=fcalc^2) { compute |Fcalc|^2 and store in Fcalc}
do phase (fcalc=0.0)
map { compute Patterson map P1 (which will be rotated)}
{ we write a hemisphere of Patterson vectors with}
extend=box { lengths less than $max_vector. }
xmin=$m_max_vector xmax=$max_vector
ymin=$m_max_vector ymax=$max_vector
zmin=0.0 zmax=$max_vector
automatic=true { use automatic scaling of map}
formatted=false
output=$p1_map { write an unformatted map file}
end
end
xrefin { make Patterson map P2 of crystal}
{===>} { unit cell for crystal}
a=67.4 b=67.4 c=290.1 alpha=90. beta=90. gamma=120.
{===>}
symmetry=(x,y,z) { operators for Patterson symmetry of crystal P622}
symmetry=(-Y,X-Y,Z)
symmetry=(Y-X,-X,Z)
symmetry=(-X,-Y,Z)
symmetry=(Y,Y-X,Z)
symmetry=(X-Y,X,Z)
symmetry=(Y,X,-Z)
symmetry=(X-Y,-Y,-Z)
symmetry=(-X,Y-X,-Z)
symmetry=(-Y,-X,-Z)
symmetry=(Y-X,Y,-Z)
symmetry=(X,X-Y,-Z)
{===>}
nreflections=20000
reflection @hexpep.xpl end { read reflections }
{===>}
resolution 20 3 { resolution range }
reduce
do amplitude ( fobs = fobs * heavy(fobs - 2.0*sigma)) { sigma cutoff }
fwind=0.1=100000
method=fft
fft
grid=0.25 { sampling grid for Patterson maps (1/4 high resol.)}
end
do amplitude (fcalc=fobs^2) { compute |Fobs|^2 and store in Fcalc}
do phase (fcalc=0.0)
map { compute Patterson map P2}
extend=unit
automatic=true { use automatic scaling of map}
formatted=false
output=$p2_map
end
end
xrefin
nrefl=10 { release some memory}
search rotation
p1input=$p1_map formatted=false
p2input=$p2_map
{===>}
range=5.0 $max_vector { Patterson vector selection for map P1}
threshold=0.0
npeaks=15000 { use 15000 largest vectors of map P1}
{===>}
tmmin=0.0 tmmax=60. { Lattman angle grid. Specify asymmetric}
t2min=0.0 t2max=90. { unit for rotation function here. See}
tpmin=0.0 tpmax=720. { Rao, S.N. et al. (1980). Acta Cryst.A36}
{ 878--884. }
delta=2.5
{ Roughly, delta should be less than ArcSin[ high resol / (3*$max_vector)].}
list=hexpep.rf { output file for cluster analysis }
nlist=6000 { analyse highest 6000 peaks of rotation function}
epsilon=0.25 { matrix norm for cluster analysis}
end
end
stop
\EOD
#
xplor <<\EOD # execute, using control data up to \EOD
remarks file xtalmr/filter.inp -- pc-refinement of rotation function peaks
parameter
@topPAR:parhcsdx.pro { Read empirical potential }
{ parameter file CHARMM19 }
end
{===>} structure @penpep.psf end { read structure file }
{===>} coor @penpep_xplor.brk { read coordinates }
evaluate ($wa=10000.) { this is the weight for the XREF energy term }
{ in this case it is arbitrary since we're not }
{ combining it with other energy term }
xrefin
{===>} { unit cell for crystal}
a=67.4 b=67.4 c=290.1 alpha=90 beta=90 gamma=120
{===>}
symmetry=(x,y,z) { operators for crystal symmetry P6(5)22}
symmetry=(-Y,X-Y,2/3+Z)
symmetry=(Y-X,-X,1/3+Z)
symmetry=(-X,-Y,1/2+Z)
symmetry=(Y,Y-X,1/6+Z)
symmetry=(X-Y,X,5/6+Z)
symmetry=(Y,X,2/3-Z)
symmetry=(X-Y,-Y,-Z)
symmetry=(-X,Y-X,1/3-Z)
symmetry=(-Y,-X,1/6-Z)
symmetry=(Y-X,Y,1/2-Z)
symmetry=(X,X-Y,5/6-Z)
SCATter ( chemical C* )
2.31000 20.8439 1.02000 10.2075 1.58860 .568700 .865000 51.6512 .215600
SCATter ( chemical N* )
12.2126 .005700 3.13220 9.89330 2.01250 28.9975 1.16630 .582600 -11.529
SCATter ( chemical O* )
3.04850 13.2771 2.28680 5.70110 1.54630 .323900 .867000 32.9089 .250800
SCATter ( chemical S* )
6.90530 1.46790 5.20340 22.2151 1.43790 .253600 1.58630 56.1720 .866900
SCATter ( chemical P* )
6.43450 1.90670 4.17910 27.1570 1.78000 0.52600 1.49080 68.1645 1.11490
SCATter ( chemical FE* )
11.1764 4.61470 7.38630 0.30050 3.39480 11.6729 0.07240 38.5566 0.97070
{===>}
nreflections=100000
reflection @hexpep.xpl end { read reflections }
{===>}
resolution 20 3 { resolution range }
reduce
do amplitude ( fobs = fobs * heavy(fobs - 2.0*sigma)) { sigma cutoff }
fwind=0.1=100000
{===>}
method=fft
fft
memory=2000000 { fft method with memory statement}
end
wa=$wa
target=E2E2 { specify target}
mbins=20 { number of bins used for E calculation}
tolerance=0. lookup=false { this makes the minimizer happy}
{ expand data to a P1 hemisphere: this sequence of}
{ statements first applies the crystal symm. ops}
{ to the current reflections. In the second step}
{ Friedel mates or other redundancies are removed.}
{ This is necessary since the application of the}
{ symmetry operators can produce Friedel mates }
{ under special conditions. }
hermitian=false expand hermitian=true symmetry reset reduce
end
flags exclude * include xref end { only use XREF energy term}
{===>} set display=hexpep.fil end { write the results of the refinement}
{ to a file called "filter.list" }
set precision="5" end
set message=off end { turn off messages and echo to reduce}
set echo=off end { output }
evaluate ($number=0)
evaluate ($counter=0)
{ loop over all orientations as specified}
{ in file rotation.rf (conventional rf)}
for $1 in ( @hexpep.rf ) loop main
evaluate ($counter=$counter+1) { this series of statements}
if ($counter=1) then evaluate($index=$1) {assigns the information of}
elseif ($counter=2) then evaluate($t1=$1) { a single line in file }
elseif ($counter=3) then evaluate($t2=$1) { rotation.rf to approp.}
elseif ($counter=4) then evaluate($t3=$1) { variables. A single line}
elseif ($counter=5) then { contains }
evaluate ($rf=$1) { $index $t1 $2 $t3 $rf. }
evaluate ($counter=0)
evaluate ($number=$number+1)
coor copy end { save current coordinates }
coor rotate euler=( $t1 $t2 $t3 ) end { and then rotate them }
{ according to the orientation}
{ specified by $t1, $t2, $t3}
energy end { compute initial energy}
evaluate ($pc1=1.0-$xref/$wa) { and store in $pc1 }
minimize rigid { rigid body minimization of the}
nstep=15 { orientation of the molecule }
drop=10.
end
evaluate ($pc2=1.0-$xref/$wa)
{ rigid body minimization of two}
{===>} { monomers. }
minimize rigid
group=( resid 3:190 or resid 299:321)
group=( resid 191:298)
drop=40.0
nstep=40
end
evaluate ($pc3=1.0-$xref/$wa)
coor swap end { fit coordinates to starting structure in}
vector do (vx=x) ( all ) { order to measure the orientation of the }
vector do (vy=y) ( all ) { PC-refined structure }
vector do (vz=z) ( all ) { the arrays vx, vy, vz are used as temp. }
coor fit end { stores in order to keep the starting }
vector do (x=vx) ( all ) { coordinates }
vector do (y=vy) ( all ) { the COOR FIT statement stores the angles}
vector do (z=vz) ( all ) { in the symbol $theta1, $theta2, $theta3 }
{ print information: orientation of rotation}
{ function peak ($t1, $t2, $t3), orientation}
{ after PC-refinement ($theta1, $theta2, }
{ $theta3), index of the rotation function,}
{ rotation function value, PCs for initial,}
{ rigid body and domain refined structures.}
display $t1 $t2 $t3 $theta1 $theta2 $theta3 $index $rf $pc1 $pc2 $pc3
end if
end loop main
stop
\EOD
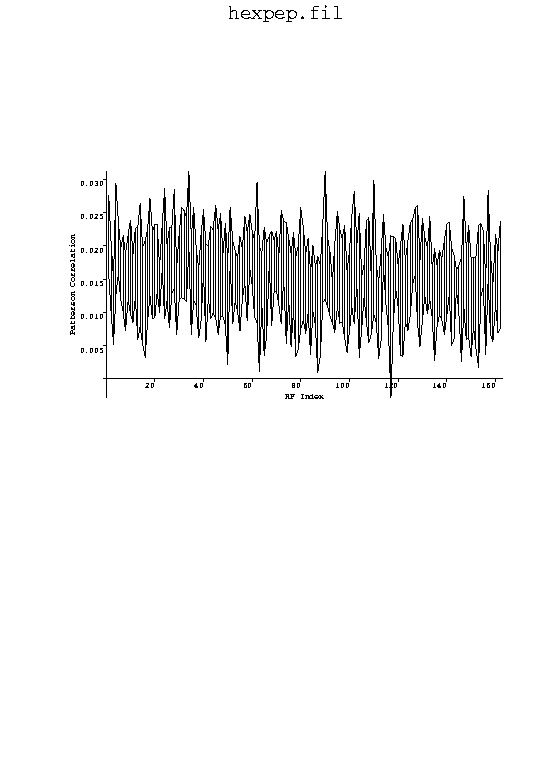
The plot shows the PC values for each RF solution before and after the segmented-body refinement. The correct solution (# 34) has the highest PC value both before and after refinement, though the refinement actually reduces the discrimination of the correct solution relative to noise solutions.
|
BACK TO INDEX | |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}