
Choosing Scaling Program (CCP4: General)
NAME
scalechoose - information about choice of scaling programThe following comes straight from a tutorial by Ian Tickle.
This document is best viewed with 12pt Times and 10pt Courier fonts.
Contents
1. Background.
Two CCP4 programs are available for determining and applying the scale factor(s) of the derivative dataset(s) relative to the reference native dataset: SCALEIT and FHSCAL. In accordance with the CCP4 philosophy of accumulating all reflection data in one file, the datasets must be contained within different columns in the same file (column-merging of files is accomplished with the MTZUTILS program).
It should be realised however that the FHSCAL program is designed specifically for derivative-to-native scaling, whereas SCALEIT is more general purpose, and can also be used for scaling of observed to calculated structure factor amplitudes. FHSCAL uses the "Kraut" scaling procedure, which is inherently more accurate than the "Wilson" and/or least squares procedure used by SCALEIT. Another difference is that SCALEIT uses one formula to fit all the scale factors, whereas FHSCAL divides the data into resolution shells, smooths the shell scale factors and then interpolates to get the final scale factor for each reflection. A third option is "local" scaling, where each reflection gets an individual scale factor which only depends on the relative scales of the reflections in its immediate neighbourhood.
Usually these differences are not important because initially only a rough scale factor is needed for the isomorphous difference Patterson, and the scale factor is refined later along with the heavy-atom parameters (i.e. 3-D coordinates, site occupancies, individual isotropic and/or anisotropic thermal parameters), and the relative overall thermal parameter for each derivative. SCALEIT has a very useful extra feature, the display of Normal probability analysis plots that can be used to decide whether the observed isomorphous and anomalous differences are really significant, or just due to errors in the measurements.
2. Scaling procedures.
The "Kraut" and "Wilson" scale factors are derived by considering the origin peak heights of the native (FP), derivative (FPH) and heavy-atom (FH) Patterson maps. Any point in a Patterson represents a vector, and the Patterson density at the point equals the sum of products of pairs of electron densities at points in the unit cell of the crystal that are separated by that vector. So the Patterson origin peak represents the sum of squares of electron densities in the unit cell. Because of the Fourier transform relationship between the Patterson and the measured intensities (= amplitude2 ), the Patterson origin peak height is simply the sum of squares of the corresponding amplitudes (this is basically Wilson's equation).
Provided the derivative structure is obtained simply by summing the native and heavy-atom structures, in other words that it is perfectly isomorphous, the derivative Patterson origin peak is just the sum of the native and heavy-atom Patterson origin peaks. Of course, the "heavy-atom structure" exists only in the imagination, as it consists only of heavy atoms in the same position as in the derivative structure, but otherwise completely empty space. Consequently we have:

Here k is the unknown scale factor needed to multiply all the measured derivative amplitudes to put them on the same scale as the measured native amplitudes. Both are of course on completely arbitrary scales, because the X-ray experiment does not take into account the incident beam intensity, crystal size, wavelength, and all the other factors that one would need to know to calculate absolute diffracted intensities. Consequently, all structure factors and occupancies in subsequent calculations are scaled relative to the arbitrarily scaled native amplitudes. This is an important point to grasp; if you don't, you will be baffled later on by occupancies greater than 1!
The heavy-atom amplitudes |FH| are of course completely unknown at this stage, and because they are on average smaller than |FP| or |FPH|, a possible assumption is simply to assume that they do not make a significant contribution and to ignore them; this gives the "Wilson" scale factor:

Alternatively, the heavy-atom amplitude can be estimated from the
isomorphous difference:
| k |FPH| -
|FP| |.
In fact, except for weak reflections where we may get
a cross-over such that
|FH| = k |FPH| + |FP|,
in the case of centric reflections (where
the phase can only take 1 of 2 values differing by 180o so the complex structure
factors are collinear), they are the same. For the remaining acentric reflections, which
are almost always the majority, because the unknown native and heavy-atom phases
are uncorrelated, it can be shown that the average isomorphous difference squared
is half the average
|FH|2.
It is of course this fact that will allow us to use the
isomorphous difference Patterson as an approximation to the
heavy-atom Patterson.
These relationships allow the unknown
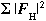 term to be eliminated, rather than
ignored, so a more accurate estimate of the scale factor
kKraut is obtained from the
resulting quadratic. For full details of the algebra, consult the FHSCAL program
documentation.
term to be eliminated, rather than
ignored, so a more accurate estimate of the scale factor
kKraut is obtained from the
resulting quadratic. For full details of the algebra, consult the FHSCAL program
documentation.
Finally, the least-squares estimate of the scale factor is obtained by minimising the
sum of weighted squares of isomorphous differences:
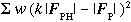 with respect to the unknown scale factor, where w is a weight equal to the reciprocal
variance of the isomorphous difference:
with respect to the unknown scale factor, where w is a weight equal to the reciprocal
variance of the isomorphous difference:
 .
However, the inherent
assumption is again that the |FH|
can be ignored;
in practice this introduces an error
of 5-10% in the scale factor, which may affect correct interpretation of the Patterson.
.
However, the inherent
assumption is again that the |FH|
can be ignored;
in practice this introduces an error
of 5-10% in the scale factor, which may affect correct interpretation of the Patterson.
To illustrate the effect of the heavy atoms on the scale factor, consider a small protein of 1000 atoms (assume for simplicity they are all N atoms). The mean scattering intensity of the protein <|FP|2> will be proportional to 1000x72 = 49000. If a single mercury atom is then introduced it will contribute 802 = 6400, so the fractional mean intensity difference between native and derivative will be 6400/49000 = 0.13.
In practice, because the introduction of the heavy atoms into the protein can anisotropically increase the disorder in the crystal, and also because of effects like absorption of X-rays by the heavy atoms, the relative scale factor can vary both with resolution and in direction, and so the procedure is a little more complicated. Programs may therefore have the option of applying an overall relative isotropic or anisotropic temperature factor to the |FPH|'s, or of applying scale factors either in equi-volume shells or in localised regions of reciprocal space.
AUTHORS
Ian Tickle, Birbeck College London, EnglandPrepared for CCP4 by Maria Turkenburg, University of York, England.